
Credit: SciTechDaily.com
Researchers at the University of Bonn examine the inner workings of machine learning applications in drug research.
Artificial intelligence (AI) has been advancing rapidly, but its inner workings often remain obscure, characterized by a “black box” nature where the process of reaching conclusions is not visible. However, a significant breakthrough has been made by Prof. Dr. Jürgen Bajorath and his team, cheminformatics experts at the University of Bonn. They have devised a technique that uncovers the operational mechanisms of certain AI systems used in pharmaceutical research.
Surprisingly, their findings indicate that these AI models primarily rely on recalling existing data rather than learning specific chemical interactions for predicting the effectiveness of drugs. Their results have recently been published in Nature Machine Intelligence.
Which drug molecule is most effective? Researchers are feverishly searching for efficient active substances to combat diseases. These compounds often dock onto protein, which usually are enzymes or receptors that trigger a specific chain of physiological actions.
In some cases, certain molecules are also intended to block undesirable reactions in the body – such as an excessive inflammatory response. Given the abundance of available chemical compounds, at a first glance this research is like searching for a needle in a haystack. Drug discovery therefore attempts to use scientific models to predict which molecules will best dock to the respective target protein and bind strongly. These potential drug candidates are then investigated in more detail in experimental studies.
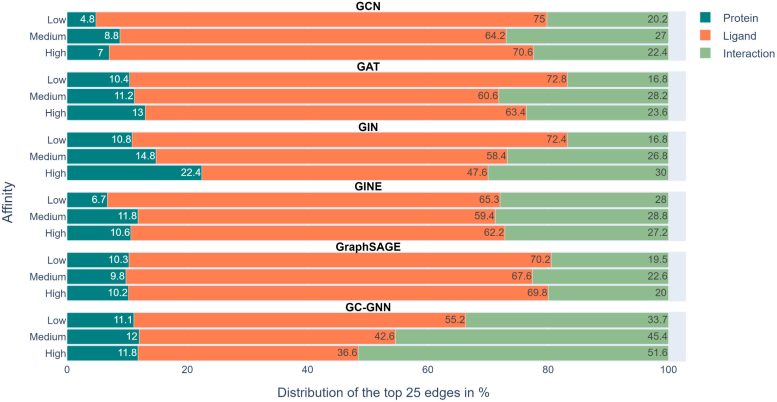
Relative proportions of edges in protein-ligand interaction graphs – determining predictions of six GNNs for different affinity subregions. The color-coded bars compare the mean proportions of protein, ligand, and interaction edges among the top 25 edges of each prediction determined with EdgeSHAPer. Credit: A. Mastropietro and J. Bajorath
Since the advance of AI, drug discovery research has also been increasingly using 
Prof. Dr. Jürgen Bajorath – from the LIMES Institute of the University of Bonn, the Bonn-Aachen International Center for Information Technology (B-IT) and the Lamarr Institute for Machine Learning and Artificial Intelligence. Credit: University of Bonn
“The GNNs are very dependent on the data they are trained with,” says the first author of the study, PhD candidate Andrea Mastropietro from Sapienza University in Rome, who conducted a part of his doctoral research in Prof. Bajorath’s group in Bonn.
The scientists trained the six GNNs with graphs extracted from structures of protein-ligand complexes, for which the mode of action and binding strength of the compounds to their target proteins was already known from experiments. The trained GNNs were then tested on other complexes. The subsequent EdgeSHAPer analysis then made it possible to understand how the GNNs generated apparently promising predictions.
“If the GNNs do what they are expected to, they need to learn the interactions between the compound and target protein and the predictions should be determined by prioritizing specific interactions,” explains Prof. Bajorath. According to the research team’s analyses, however, the six GNNs essentially failed to do so. Most GNNs only learned a few protein-drug interactions and mainly focused on the ligands. Bajorath: “To predict the binding strength of a molecule to a target protein, the models mainly ‘remembered’ chemically similar molecules that they encountered during training and their binding data, regardless of the target protein. These learned chemical similarities then essentially determined the predictions.”
According to the scientists, this is largely reminiscent of the “Clever Hans effect”. This effect refers to a horse that could apparently count. How often Hans tapped his hoof was supposed to indicate the result of a calculation. As it turned out later, however, the horse was not able to calculate at all, but deduced expected results from nuances in the facial expressions and gestures of his companion.
What do these findings mean for drug discovery research? “It is generally not tenable that GNNs learn chemical interactions between active substances and proteins,” says the cheminformatics scientist. Their predictions are largely overrated because forecasts of equivalent quality can be made using chemical knowledge and simpler methods. However, the research also offers opportunities for AI. Two of the GNN-examined models displayed a clear tendency to learn more interactions when the potency of test compounds increased. “It’s worth taking a closer look here,” says Bajorath. Perhaps these GNNs could be further improved in the desired direction through modified representations and training techniques. However, the assumption that physical quantities can be learned on the basis of molecular graphs should generally be treated with caution. “AI is not black magic,” says Bajorath.
Even more light into the darkness of AI
In fact, he sees the previous open-access publication of EdgeSHAPer and other specially developed analysis tools as promising approaches to shed light on the black box of AI models. His team’s approach currently focuses on GNNs and new “chemical language models.”
“The development of methods for explaining predictions of complex models is an important area of AI research. There are also approaches for other network architectures such as language models that help to better understand how machine learning arrives at its results,” says Bajorath. He expects that exciting things will soon also happen in the field of “Explainable AI” at the Lamarr Institute, where he is a PI and Chair of AI in the Life Sciences.
Reference: “Learning characteristics of graph neural networks predicting protein–ligand affinities” by Andrea Mastropietro, Giuseppe Pasculli and Jürgen Bajorath, 13 November 2023, Nature Machine Intelligence.DOI: 10.1038/s42256-023-00756-9